
School of Computer Science, Carnegie Mellon University
Hi! I am an incoming Ph.D. student in Computational Biology (CPCB) at Carnegie Mellon University, affiliated with the School of Computer Science. My research is motivated by a central question: how can we extract reliable biological insight from noisy and incomplete data?
During my military service as a medic, I witnessed how clinical decisions were often made under uncertainty due to limited and imperfect data. This experience led me to pursue computational approaches, adding a second major in Computer Science to build a rigorous foundation in algorithms and statistical modeling.
At the University of North Carolina at Chapel Hill, I worked in Prof. Terry Fureys lab on epigenomic method development. There, I developed WAD, a wavelet-based framework for bulk ATAC-seq deconvolution that addresses the intrinsic sparsity and noise of chromatin accessibility data. More broadly, I am interested in building robust and interpretable computational methods for understanding gene regulation from complex genomic data.
Warning
Problem: The current name of your GitHub Pages repository ("Solution: Please consider renaming the repository to "
http://".
However, if the current repository name is intended, you can ignore this message by removing "{% include widgets/debug_repo_name.html %}" in index.html.
Action required
Problem: The current root path of this site is "baseurl ("_config.yml.
Solution: Please set the
baseurl in _config.yml to "Education
-
 Carnegie Mellon UniversityPh.D. in Computational Biology
Carnegie Mellon UniversityPh.D. in Computational Biology
School of Computer ScienceStarting Aug. 2026 -
 University of North Carolina at Chapel HillStudent Exchange ProgramJan. 2025 - Jul. 2025
University of North Carolina at Chapel HillStudent Exchange ProgramJan. 2025 - Jul. 2025 -
 Korea UniversityB.S. in Life Sciences / Computer Science and EngineeringMar. 2020 - Feb. 2026
Korea UniversityB.S. in Life Sciences / Computer Science and EngineeringMar. 2020 - Feb. 2026
Experience
-
University of North Carolina at Chapel HillResearch Assistant - Terry Furey's labJan. 2025 - Present
-
 Republic of Korean ArmyMedic (Sergeant)Oct. 2021 - Mar. 2023
Republic of Korean ArmyMedic (Sergeant)Oct. 2021 - Mar. 2023
Honors & Awards
-
Korea-US Advanced Industry Scholarship Recipient2025
-
1st (1/400), Military Physical Fitness Test2021
Selected Publications (view all )
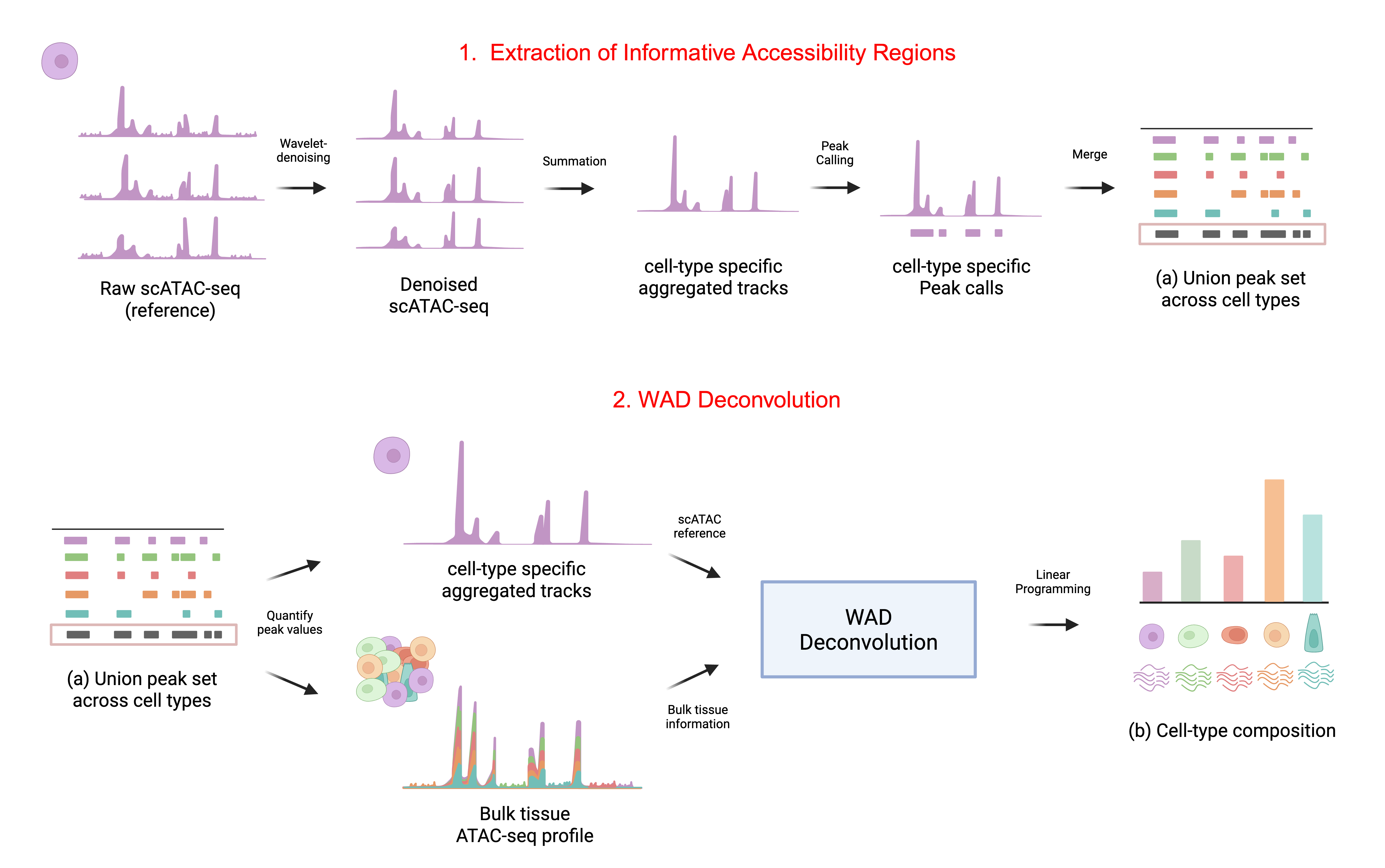
WAD: a wavelet-based linear programming method using L1-minimal reconstruction loss for accessible chromatin data deconvolution
Jeongho Chae, Benjamin McMichael, Terrence S. Furey
Preprint 2025
We present WAD (Wavelet-based Accessible chromatin Deconvolution), a principled framework for robust estimation of cell type composition of bulk accessible chromatin data such as from the ATAC-seq assay. To determine informative reference cell profiles from single-cell accessible chromatin studies, WAD leverages wavelet-based denoising to suppress stochastic noise while preserving local chromatin continuity. Cell type proportion inference is reformulated as an L1-minimal linear programming problem, enabling scalable and interpretable solutions. Across 700 in silico pseudo-bulk mixtures generated from single-cell data, WAD achieved a consistently lower mean absolute error (MAE) and higher concordance (r > 0.85) than existing machine learning-based methods. These results demonstrate that wavelet-based feature extraction provides a biologically grounded and computationally efficient approach to chromatin signal deconvolution.
WAD: a wavelet-based linear programming method using L1-minimal reconstruction loss for accessible chromatin data deconvolution
Jeongho Chae, Benjamin McMichael, Terrence S. Furey
Preprint 2025
We present WAD (Wavelet-based Accessible chromatin Deconvolution), a principled framework for robust estimation of cell type composition of bulk accessible chromatin data such as from the ATAC-seq assay. To determine informative reference cell profiles from single-cell accessible chromatin studies, WAD leverages wavelet-based denoising to suppress stochastic noise while preserving local chromatin continuity. Cell type proportion inference is reformulated as an L1-minimal linear programming problem, enabling scalable and interpretable solutions. Across 700 in silico pseudo-bulk mixtures generated from single-cell data, WAD achieved a consistently lower mean absolute error (MAE) and higher concordance (r > 0.85) than existing machine learning-based methods. These results demonstrate that wavelet-based feature extraction provides a biologically grounded and computationally efficient approach to chromatin signal deconvolution.